Velg et land eller en region


-
Argentina (Español)

-
Australia (English)

-
Austria (Deutsch)

-
Bahrain (العربية)

-
Bahrain (English)
-
België (Nederlands)

-
Belgique (Français)
-
Brazil (Portugues)

-
Canada (English)

-
Canada (Français)
-
Chile (Español)

-
Colombia (Español)

-
Croatia (Croatian)

-
Denmark (Danish)

-
Deutschland (Deutsch)

-
Europe (English)

-
France (Français)

-
Greece (Ελληνικά)

-
Italia (Italiano)

-
Hungary (Magyar)

-
Lietuva (Lietuviškai)

-
Mexico (Español)

-
日本 (日本語)

-
대한민국 (한국어)

-
Kuwait (العربية)

-
Kuwait (English)
-
Nederland (Nederlands)

-
Norge (Norsk)
-
Oman (العربية)

-
Oman (English)
-
Polska (Polskie)

-
Portugal (Portuguese)

-
Qatar (العربية)

-
Qatar (English)
-
Saudi Arabia (العربية)

-
Saudi Arabia (English)
-
Slovakia (Slovak)

-
Slovenia (Slovenščina)

-
Spain (Español)

-
Suomi (Suomi)

-
Sverige (Svenska)

-
Schweiz (Deutsch)

-
台灣 (中文)

-
United States (English)

-
UAE (العربية)

-
UAE (English)
Personene som vises her er virkelige pasienter, og det nødvendige samtykket for å bruke historiene deres er innhentet både fra pasientene og fra familiene deres. Bildene er kun for illustrasjon.


HVA ER SMA
HVA FORÅRSAKER SMA?
Spinal muskelatrofi (SMA) er en sjelden nevromuskulær sykdom forårsaket av en mutasjon i survival motor neuron 1-genet (SMN1) 1


HVA ER SMA
TYPER AV SMA
SMA er delt inn i ulike typer i henhold til alder for sykdomsdebut og funksjonsevne. Det finnes også ulike alvorlighetsgrader innen hver type.2


HVA ER SMA
HVORDAN BLIR SMA NEDARVET?
SMA er en nedarvet sykdom, og i motsetning til mange andre sjeldne nevromuskulære sykdommer er det en klar forståelse av den spesifikke årsaken til spinal muskelatrofi.3

HVA ER SMA
SYKDOMMER MED LIGNENDE SYMPTOMER
Selv om SMA kjennetegnes av visse tegn og symptomer, er det andre sykdommer som gir de samme symptomene, men med andre genetiske årsaker.
HVA ER SMA
DIAGNOSTISERING OG TESTING
Hvis det foreligger mistanke om SMA, er det viktig at den innledende diagnosen blir bekreftet med genetisk testing.4


HVA ER SMA
MYTER OG FAKTA
Vi har samlet en del fakta rundt spinal muskelatrofi (SMA) som vi håper kan bidra til en bedre forståelse av sykdommen.

HVA ER SMA
HVA FORÅRSAKER SMA?
Spinal muskelatrofi (SMA) er en sjelden nevromuskulær sykdom forårsaket av en mutasjon i survival motor neuron 1-genet (SMN1)1

HVA ER SMA
TYPER AV SMA
SMA er delt inn i ulike typer i henhold til alder for sykdomsdebut og funksjonsevne. Det finnes også ulike alvorlighetsgrader innen hver type.2

HVA ER SMA
HVORDAN BLIR SMA NEDARVET?
SMA er en nedarvet sykdom, og i motsetning til mange andre sjeldne nevromuskulære sykdommer er det en klar forståelse av den spesifikke årsaken til spinal muskelatrofi.3

HVA ER SMA
SYKDOMMER MED LIGNENDE SYMPTOMER
Selv om SMA kjennetegnes av visse tegn og symptomer, er det andre sykdommer som gir de samme symptomene, men med andre genetiske årsaker.
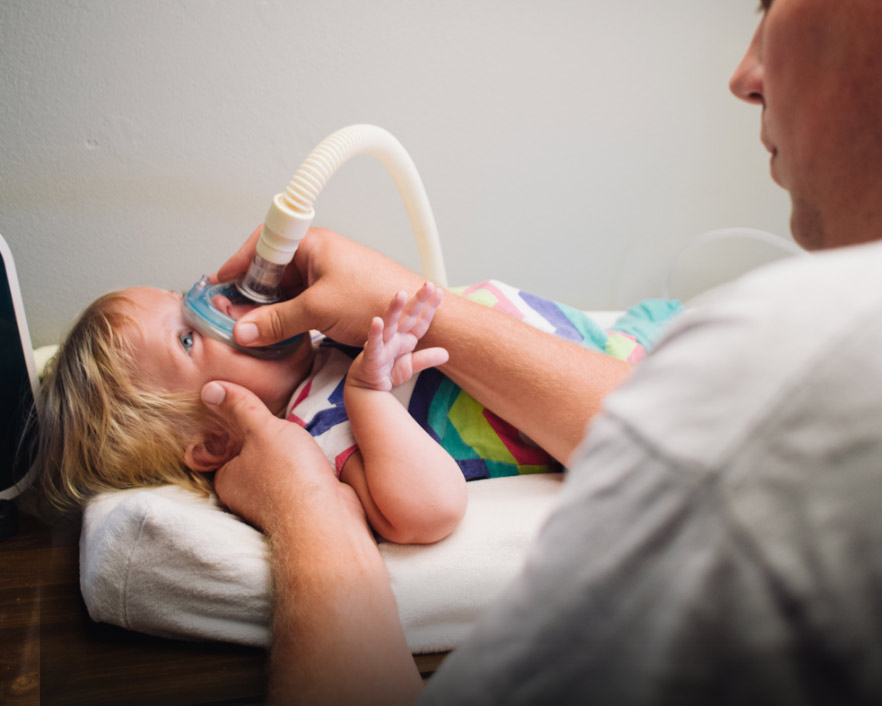
HVA ER SMA
DIAGNOSTISERING OG TESTING
Hvis det foreligger mistanke om SMA, er det viktig at den innledende diagnosen blir bekreftet med genetisk testing.4

HVA ER SMA
MYTER OG FAKTA
Vi har samlet en del fakta rundt spinal muskelatrofi (SMA) som vi håper kan bidra til en bedre forståelse av sykdommen.
Personene som vises her er virkelige pasienter, og det nødvendige samtykket for å bruke historiene deres er innhentet både fra pasientene og fra familiene deres. Bildene er kun for illustrasjon.
References
1. Lunn MR, Wang CH. Spinal muscular atrophy. Lancet. 2008;371(9630):2120-2133.
2. Kolb SJ, Kissel JT. Spinal muscular atrophy. Arch Neurol. 2011;68(8):979-984.
3. National Organization for Rare Disorders. Spinal Muscular Atrophy. [online] 2012 [cited 2020 Sep 22].
Available from: URL: https://rarediseases.org/rare-diseases/spinal-muscular-atrophy/.
4. Mercuri E, et al. Diagnosis and management of spinal muscular atrophy: Part 1: Recommendations for diagnosis, rehabilitation, orthopedic and nutritional care. Neuromuscul Disord 2018;28(2):103-115.